
Platform Leader: Jared Cole
Deputy Platform Leader: Salvy Russo
Researchers
International Collaborations
Exciton Science Research Groups
Exciton dynamics in materials and across hybrid interfaces are highly complex and are dependent on nanoscale morphology, spin polarisation and vibrational properties. The aim of this platform is to develop computational modelling techniques to understand the exciton dynamics in single molecules or organic-inorganic hybrid systems.
In this platform, we employ a combination of different theoretical approaches to examine the interplay between electronic coupling, spin and structure on exciton diffusion and delocalisation in disordered organic and hybrid materials on scales ranging from the atomistic to the device level. To this end, this platform will develop multiscale computational methods that can help interpret and predict exciton behaviour from the nanoscale to the device level.
| Name | Node |
|---|---|
| Salvy Russo | RMIT |
| Alison Funston | Monash |
| Jared Cole | RMIT |
| Paul Mulvaney | UoM |
| Trevor Smith | UoM |
| Girish Lakhwani | USyd |
| Dane McCamey | UNSW |
| Asaph Widmer-Cooper | USyd |
| Name | Node |
|---|---|
| Kavan Modi | Monash |
| Ivan Kassal | USyd |
| Name | Node |
|---|---|
| Mykhailo Klymenko | RMIT |
| Igor Lyskov | RMIT |
| Robert Shaw | RMIT |
| Nastaran Meftahi | RMIT |
| Francesco Campaioli | RMIT |
| Yawei Liu | USyd |
| Stefano Bernardi | USyd |
| Martin Cyster | RMIT |
| Jesse Collis | UoM |
| Jesse Vaitkus | RMIT |
This platform is primarily concerned with method development, which is led by postdoctoral researchers. The postgraduate students using these methods are reported under the other platforms, which focus on the applications of the methods developed within this platform.
As this is a central fundamental theme, exciton transport insights and multiscale modelling provides guidance across all platforms and capabilities, most particularly within Theme 1 and 2. The first three years involved a large amount of expertise and capability development. We are now applying these techniques to open problems of interest to the Centre, which should see an increase in cross-nodal projects and publications.
RMIT/UNSW (Cole, Russo, Schmidt) has been successful in obtaining funding of a standard proposal #6508, "Ab initio study of hybrid inorganic-organic semiconductor interfaces for photovoltaic applications", through the Molecular Foundry. The proposal plans to study the interface between Silicon (Si) and a singlet fission material like tetracene using advanced quantum chemistry techniques recently developed at the Molecular Foundry. Progress to date is steady and we have been invited to submit a continuation request in the March/April 2021 round.
A key benefit of the Molecular Foundry proposal is training in the use of the BerkeleyGW code which was developed at Lawrence Berkeley National Laboratory (LBNL), which complements our existing expertise (Russo) in VASP+GW. These techniques allow accurate calculation of the excited state properties of materials which is essential for computing exciton levels and binding energies. We have also made good progress in matching quantum chemistry techniques with open-quantum systems techniques to study the movement of excitons in both polymer and crystalline materials.
Results published in 2020 which demonstrate or take advantage of our multiscale modelling capabilities include:
Other important results and progress in 2020 include:
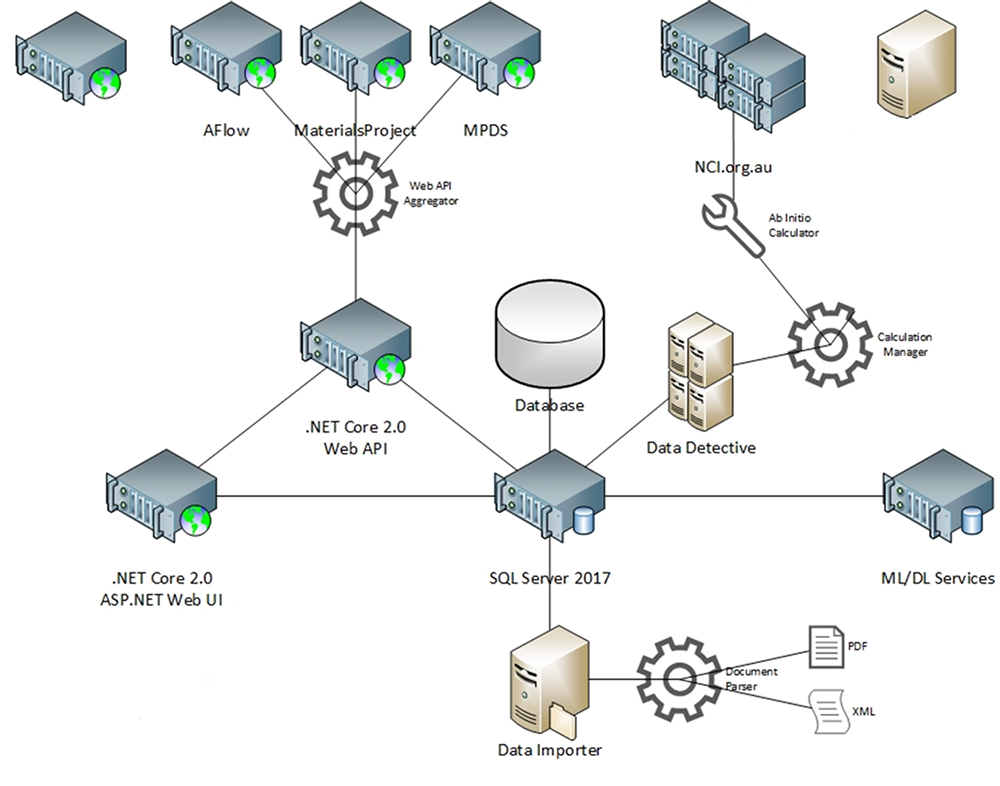
Schematic diagram of the Materials Discovery Software Toolkit, which enables data curation from International Materials Science Databases and Machine Learning Prediction of data.
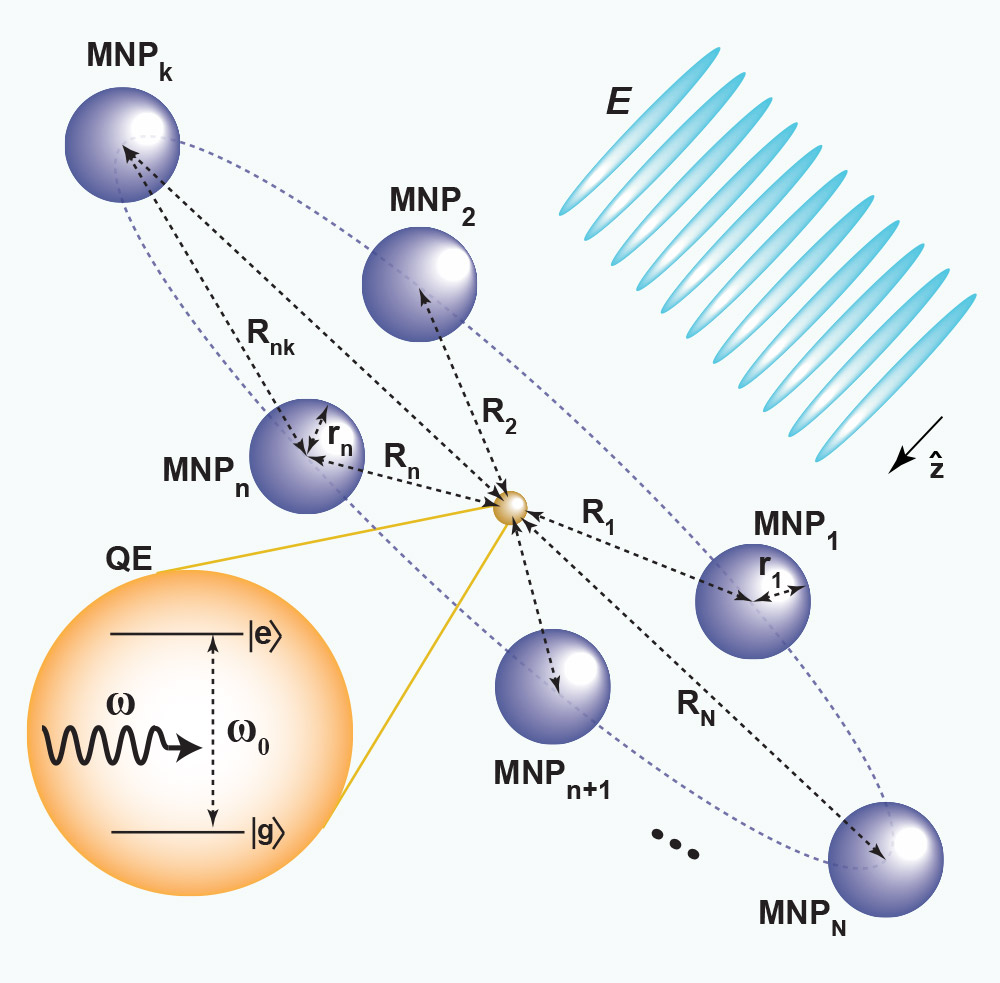
Schematic diagram of a quantum dot surrounded by metallic nanoparticles. When applying a laser field to the quantum dot, the nanoparticles dramatically modify the optical response of the dot.
Dr Liang Tan, Molecular Foundry, Lawrence Berkeley National Laboratory, USA
Collaboration with Mike Klymenko et al. on applying GW methods to exciton stability and energy levels at organic/inorganic interfaces (Si/Tc).
Staffing: Recruitment has been difficult over the last few years due to a shortage of qualified applicants and relatively inflexible University regulations. In late 2019 we were finally able to offer all the necessary appointments. Unfortunately, two of these appointments were for internationals requiring visas. These visas were then swept up in the COVID related visa freeze. Based on the delay, the University has rescinded the offers. However, there is no plan to re-advertise as the current team has the required skill set and therefore the funding will be invested in contract extensions beyond the original two or three-year contracts.
Device Modelling: In previous ISAC and internal reviews, a weakness was identified in both device modelling and transport modelling at micron to millimetre scales. This year, a “joint postdoc” appointment has been made between RMIT (Russo) and Monash (Jasieniak) to focus on device models for current and future planned experiments at Monash.
In 2021, a range of modelling techniques will continue to be explored and benchmarked. In addition, there are an increasing number of projects underway using existing techniques to model experimental systems within the Centre and with collaborators.
Areas of interest within the platform moving forward: